SeedScan - Data files
SeedScan requires several input files to generate the resulting data matrix.
Just drag the files from Windows Explorer into the respective text field,
or click the "..." button to select a file.
- Barcodes - a list of barcodes sequences used for multiplexing
- Barcode pairs - a list of bardcode pairs used in forward/reverse read to identify a specific multiplex sample
- Seeds - a list of (gene) target sequences
- Sequences - Sequence files from NGS sequencing
- Result matrix - A tab-delmited text file containig counts for all seeds/samples
Barcodes
During amplification of the specific sample, a pair of sample specific PCR-primers is used to introduce sample specific "barcode" sequences.
SeedScan expects a barcode list:
- Tab delimited text file
- First line = Header => not used
- Empty lines => ignored
- Lines starting with "#" = any comment => not used
- all other lines are interpreted as barcodes
- one barcode per line
- Tab delimited: barcode name and barcode sequences
-
- Barcode should ONLY contain base characters (A,C,G,T), not case sensitive.
No additional/leading/tailing spaces, base numbering, ...
To preview the barcode file, click the View button right to the Barcode field:
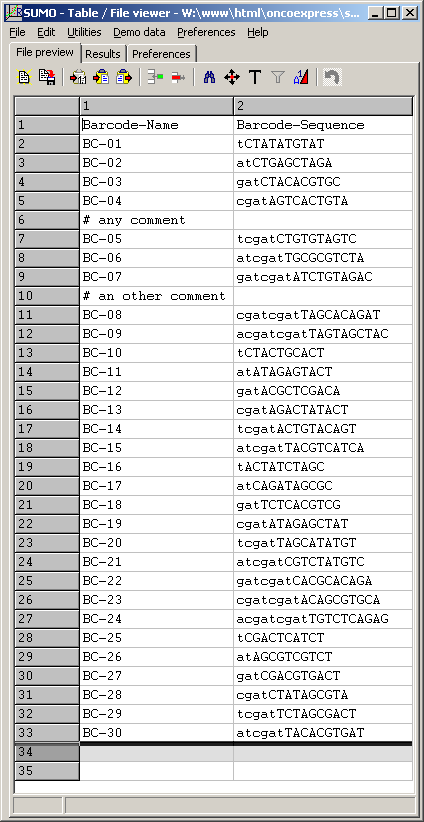
(Click image to see the file)
Obviously, the barcode list may be applied to any analysis where the same barcodes were used.
The barcode list should not contain more barcodes than used for the multiplex PCRs.
Although they should not disturb the counting, many additional (not used) barcodes would slow down the analysis.
Barcode pairs
To improve the identification of multiplex samples, a forward/reverse barcode may be introduced during PCR amplification.
Thus a list of "allowed" barcode-pairs is required.
In cases where multiple seed libraries were used to generate the individual sample, additionlally specify the ID of the respective seed-library.
SeedScan expects a barcode-pair list:
- Tab delimited text file
- First line = Header => not used
- Empty lines => ignored
- Lines starting with "#" = any comment => not used
- all other lines are interpreted as barcode-pairs
- one barcode-pair per line
- Tab delimited: barcode name (muliplex samplename), ID-Barcode1, ID-Barcode2, Seed library-ID
- Barcode-IDs relative to above barcode list.
First barcode = 2. line in file (1. = header), omit any/empty comment lines.
- Seed library-ID relative to the seed library files defined.
Either 1,2,3,... or A,B,C,...
In the example Gecko Library A and B were used. Thus A or B are defined to scan the sample to its respective library.
1/A corresponds to the first library file added to the Seed library text field, 2/B to the second library file, ....
To preview the barcode-pairs file, click the View button right to the Barcode-pairs field:
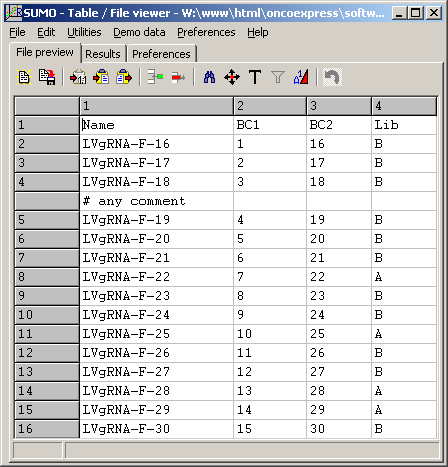
(Click image to see the file)
The barcode-pairs file has to be adapted to a specific analysis.
Check that:
- Correct Bar code lists are used
- Correect bar-code pairs are defined.
Keep in mind: empty/comment lines in bar code lists are ignored and not counted !!
E.g.: Barcode pair LVgRNA-F-20 (line six in the eample above references
Barcode pair 5<=>20 from above bar code list.
Due to comment lines BC-05 is found in line 7 of the bar code file,
BC-20 in line 23 of the respective barcode list file !
- Check that Library IDs correspond to the order how the library files are defined.
"A","B","C",... or "1","2","3", ... only give the number of the specified library
but do not reflect any internal naming of libraries.
Seeds
The sequence are compared to the list of (gene) target specific constructs used in the experiment.
SeedScan expects a list of these seeds:
- Tab delimited text file
- First line = Header => not used
- Empty lines => ignored
- Lines starting with "#" = any comment => not used
- all other lines are interpreted as (gene) target specific seed sequences
- one seed sequence per line
- Tab delimited: Target name and seed sequence
- Seeds should ONLY contain base characters (A,C,G,T), not case sensitive.
No additional/leading/tailing spaces, base numbering, ...
- All seeds start at the same position in all constructs
- All seeds have the same length
To preview the seeds file, click the View button right to the Seed file field:
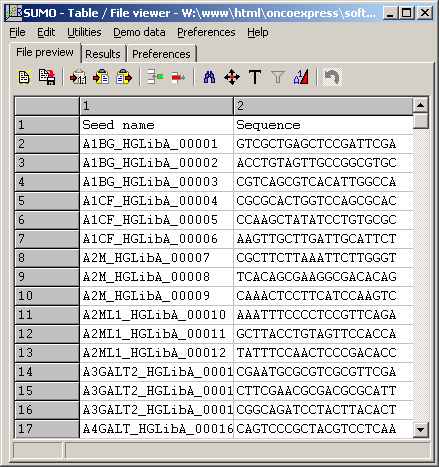
(Click image to see the file)
The example shows a part (first hundred from 65383 seeds) of the Gecko A library.
Sequences
Sequences to analyze (one forward and optional one reverse read) are multiple sequence files in FastQ sequence format.
- ASCII text files
- Four lines per read
- First line: sequence name, coordinates of the respective cluster, ... => not used
- Second line: Sequence
100.. 250 base characters (A,C,G,T) non case sensitive
No additional/leading/trailing spaces, base numbering, ...
- Thrid line: orientation => not used
- Fourth line: Base quality string => not used
- Forward/reverse read FastQ file contain the same number of reads in exactly the same order.
FastQ files may be supplied as straight text files or as GZ-compressed archives.
The example shows a part of a FastQ multiple sequence file, sequence lines highlighted.:
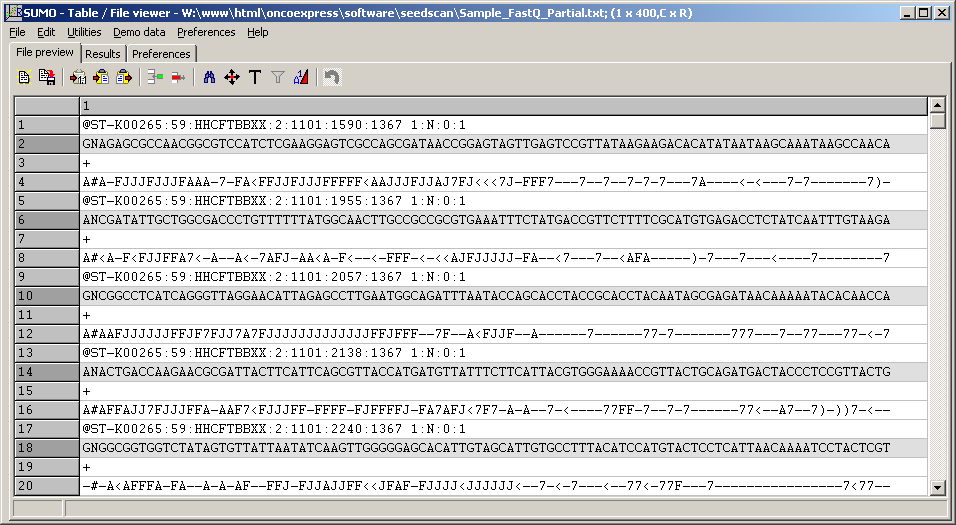
(Click image to see the file)
First hundred from ~350 Million reads in the original file.
Result matrix
Counting results are saved as a tab-delimited count matrix.
- tab delimited text
- samples (barcode-pairs) in columns
- target (gene) specific seeds in rows
- several annotation columns
Seed name, Seed sequence, Total count across all samples, Relative proportion of this seed, ...
- several annotation rows
Sample (barcode-pair)name, barcode sequences, total count accross all seeds, Relative proportion of this sample
The matrix is stored at regluar intervals during an anlysis run. Thus the scan may be Paused and the result matrix may be viewed
and preliminaryly analyzed.
The TotCount row gives the absolute number od sequences for the respective barcode-pair / sample.
This allows to compare total amount of analyzed DNA and thus, draw conclusions about sample concentration/preparation.
To allow easy comparison of samples from different runs, count values for the seeds (genes) are normalized to 1 Million total reads (~RPFM).
As each seed should match only once on a gene, counts are not normalized to the target (gene) length.
To preview the result matrix file, click the View button right to the Result matrix field:
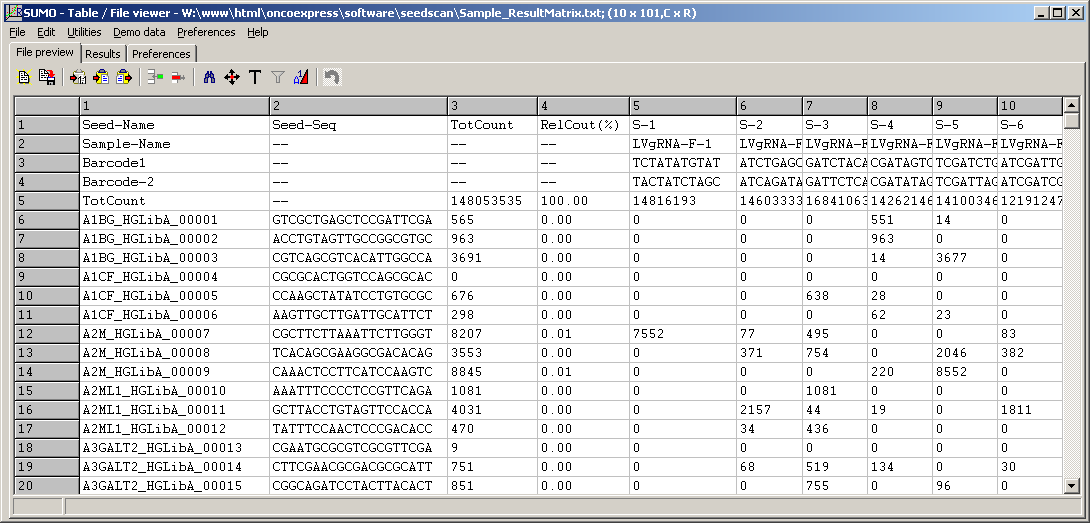
(Click image to see the file)
The example shows a part (first hundred from 65383 seeds, 6 from 15 samples) of a targeting screen with the Gecko A library.
Due to the experiments selection pressure only few target's guides are preserved in the analyzed cells. Thus most counts seed/sample-cells are 0.
Especially in the tiny partial result sample.