

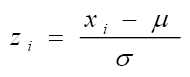
Bolstad, B. M., Irizarry R. A., Astrand, M, and Speed, T. P. (2003)especially cited unpublished manuscript
A Comparison of Normalization Methods for High Density Oligonucleotide Array Data Based on Bias and Variance.
Bioinformatics 19,2,pp 185-193
Bolstad, B (2001) Probe Level Quantile Normalization for High Density Oligonucleotide Array Data
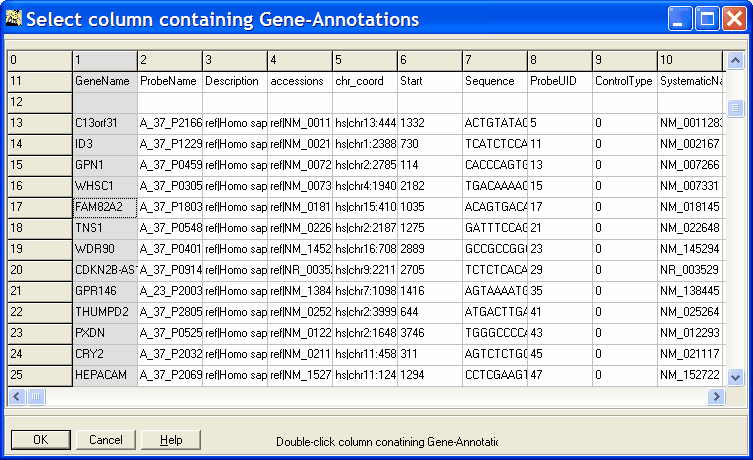

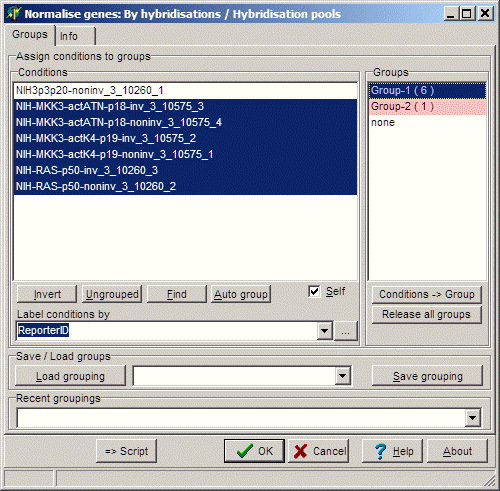
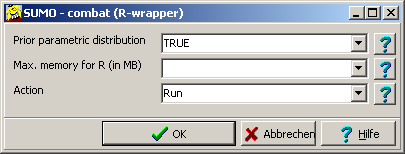
library(SVA)In case of error messages, install SVA.
# old Bioconductor versions:
# source("http://bioconductor.org/biocLite.R")
# biocLite("DESeq2")
#
For actual Bioconductor verions use:
BiocManager::install("DESeq2")
install.packages("sva")
and try again to load SVA.
http_proxy=http://your-proxyserver-name:80
https_proxy=http://your-proxyserver-name:80
Obviously you shold replace "your-proxyserver-address" with your organization 's proxyserver name and port
library("DESEQ2")
In case of error messages, install DESEQ2.
# old Bioconductor versions:
# source("http://bioconductor.org/biocLite.R")
# biocLite("DESeq2")
#
For actual Bioconductor verions use:
BiocManager::install("DESeq2")
install.packages("BiocManager")
and try again to load DESEQ2.
http_proxy=http://your-proxyserver-name:80
https_proxy=http://your-proxyserver-name:80
Obviously you shold replace "your-proxyserver-address" with your organization 's proxyserver name and port
library("DESeq2")
In case of error messages, install DESEQ2.
# old Bioconductor versions:
# source("http://bioconductor.org/biocLite.R")
# biocLite("DESeq2")
#
For actual Bioconductor verisons use:
BiocManager::install("DESeq2")
install.packages("BiocManager")
and try again to load DESeq2.
http_proxy=http://your-proxyserver-name:80
https_proxy=http://your-proxyserver-name:80
Obviously you shold replace "your-proxyserver-address" with your organization 's proxyserver name and port.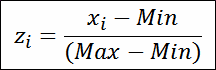
cv = ( 1 + 1/4n ) * s / m | |
| with: s=sample standard deviation, m=sample mean, n=sample size more details |
CQD = ( Q3 - Q1 ) / (Q3 + Q1 ) | |
| with: Q1=value of first quartiles, value of (n+1)/4th item Q3=value of third quartile: value of (3*(n+1))/4th item more details |