| Subject name |
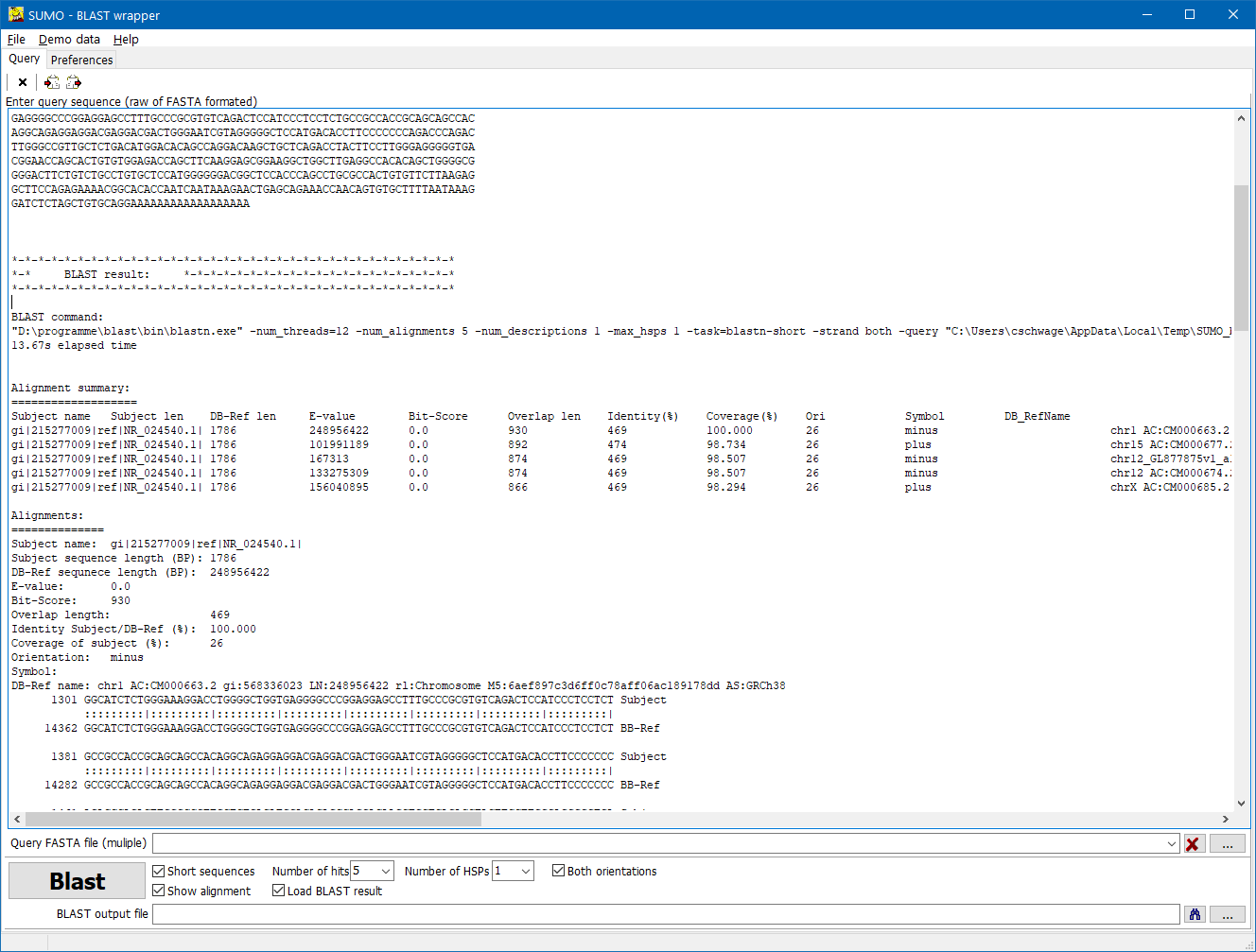
Name of the sequence, as extracted from the FASTA header line |
| Subject length |
Length of the search sequence |
| DB-Ref len |
Length of the matched sequence in the reference data base. |
| E-value |
Expect value, indicating how probable it is to get such an alignment.
E-values close or larger 1 indicate a non specific just by chance expected alignment.
Very small E-values indicate non random specific alignments.
The E-value depends obviously on length and quality of the alignment between search and data base sequence.
But databse size as well as search sequence length will influence the E-value. |
| Bit score | |
| Overlap length |
Contiguous part of the search sequence which matches the respective bata base sequence.
Ideally, Overlap length should be equal to Subject length. |
| Identity(%) |
Fraction of the search sequence which matches the data base sequence.
Ideally, Identity should be 100, i.e. search and data base sequence are identical. |
| Coverage |
Fraction of the search sequence which matches the data base.
Ideally Coverage should be 100, i.e. the complete search sequence matches the data base. |
| Ori | Orientation of the search sequence in respect to the data base |
| Symbol |
Gene symbol, extracted from the data base sequence |
| DB-RefName | Name of the matched sequence in the reference data base. |
Peferences
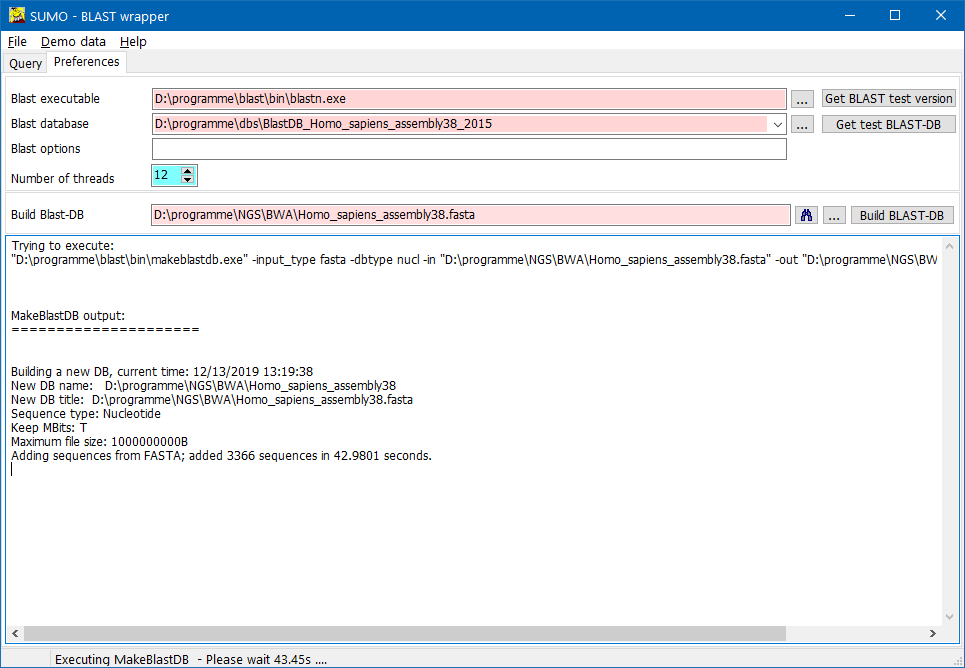
On the Preferences page, define location of BLAST executable and databases:
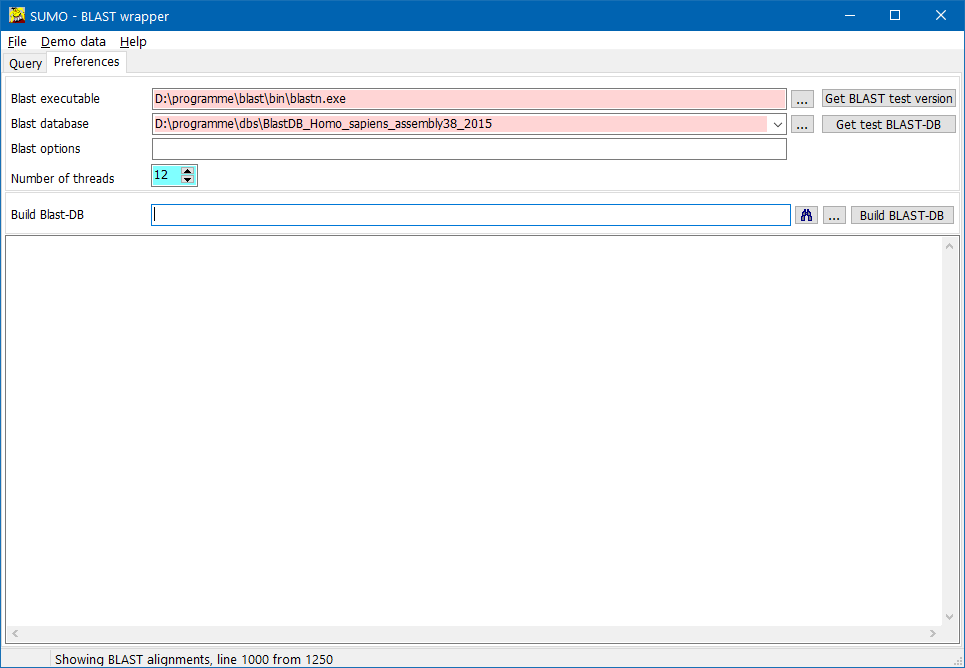
Click the ... buttons to open a file selection dialog for the respective file.
Alternatively, drag a file from Windows explorer into the respective text field.
- BLAST executable - the blast program file Required !!
(eg. "blastn.exe" for search of nucleotide sequneces against a nucleotide database)
Download BLAST package from NCBI's ftp
site.
(e.g. ncbi-blast-x.x.x+-win64.exe.
Execute the self extracting BLAST installer.
Just for testing you may download an older BLAST package version from SUMO site and place into ..\SUMO-programfolder\BLAST\ and setup blastn.exe,
but better get a present version from NCBI.
Instead of blastn.exe you may select any of the BLAST versions delivered with NCBI's BLAST package:
| Program | Query | Data base | |
| blastn | DNA | DNA | |
| blastp | Protein | Protein | |
| blastx | DNA | Protein | compare the six-frame DNA translation against protein |
| tblastn | Protein | DNA | compare protein query sequence against six frame translated DNA |
| tblastx | DNA | DNA | compare six frame translation DNA against six-frame translation DNA |
- BLAST database - a Blast transformed/formated reference sequence data base. Required !!
The BLAST database consists of several files. Just define the main part of the file.
E.g. When building a BLAST-DB form all Human_RefSeq_mRNAs, you will get three BlastDB files:
"Human_RefSeq_mRNA.nhr, Human_RefSeq_mRNA.nin, Human_RefSeq_mRNA.nsq".
Thus specify as BLAST-DB name: "Human_RefSeq_mRNA". Blast will find and use all required files.
Either dwonload pre-build data bases e.g. from NCBI,
or build the data base yourself with the "makeblastdb.exe" tool (contained in the NCBI blast package) starting with FASTA sequence files (see below).
Just for testing you may download older BLAST databases for Human RefSeq RNA / HUman Genome 38 from SUMO site and place into SUMO-programfolder\BLAST\.
- Number of threads - As more as faster BLAST will run.
To allow parallel interactive work while blast is running it may be recommended to define less threads for BLAST as hyperthreads/cores are in your computer.
E.g. with a Quad-core I7-CPU, you will have 8 hyperthreads: define 6 threads for BLAST. The remaining 2 Hyperthreads will ensure convenient inter-active work.
With a Quad-Core I5-CPU, you will have 4 threads: define 3 threads for BLAST.
With Dual-Core I3-CPU, you will have 2 threads: define 1 for BLAST
More BLAST-threads as Cores/Hyperthread in your system are not helpful.
Build BLAST-DB
It may be recommened to generate a new or update the BLAST reference database from time to time.
One way may be, to download reference sequence files (multiple FASTA format) e.g. from NCBI and build the data base on your system.
If you have a Multiple FASTA file containing your reference sequences, you may just select this mFASTA-file for the Build BLAST-DB text field.
Now click the Build-BLAST-DB button.
SUMO will lauch the "makeblastdb.exe" utility from the installed blast package to generate a BLAST-DB.
The newly generated BLAST-DB is saved into the folder where the selected mFASTA file is found.
Processing success-/ error-messages are shown in the text box on the Preferences-tabsheet.
If you want to regularly update your BLAST-DBs you might create a Windows script and run it (e.g. once per month) with the Windows scheduler.
Such a Windows script file might look like:
rem
rem get RefSEQ mRNA fasta files from NCBI ftp-site and convert into BlastDB
rem V1.00a, from 31.03.2017, c.schwager[at]dkfz.de
rem
rem get files from ncbi via ftp, using ftp command file ftp.txt
ftp -s:ftp.txt
rem
rem unpack all just downloaded archives
c:\programme\7-zip\7z.exe -y e *.gz
rem
rem append all just unpacked fasta file into one
copy *.fna Human_RefSeq_mRNA.fas
rem
rem build blast database
d:\programme\blast\bin\makeblastdb.exe -input_type fasta -dbtype nucl -in Human_RefSeq_mRNA.fas -out BlastDB\Human_RefSeq_mRNA
rem
rem thats it
pause
- Download respective DNA sequence files from NCBI's ftp site, using MS Windows ftp-command line tool.
All ftp commands are combind in a ftp script file (ftp.txt, see below).
- unpack all downloaded gzipped files, using e.g. the free tool 7Zip
- Copy/append all extracted sequence files into one (Human_RefSeq_mRNA.fas)
- Use the makeblastdb.exe tool to build a Blast tranformed database from the fasta sequence file.
makeblasdb.exe is a part of the Blast package donwloadable from NCBI
Obviously, you have to modify any file/program locations according to your sytem.
Also, references to locatons at NCBI may change over time.
The ftp script might look like:
open ftp.ncbi.nih.gov
anonymous
yourname@yourinstitue.org
cd refseq
cd H_sapiens
cd mRNA_Prot
bin
hash
prompt no
mget human*rna.fna.gz
close
bye
- open ...: Connect to the respective ftp server
- anonymous: use username "anonymous"
- xxxx@yyyyyorg: as password, use your e-mail address
- cd ....: change to folders where your desired sequence files are found
-
- bin: set binary transfer mode - we are downloading compressed files
- hash: enable download progress indication
- prompt no:suppress any interactive confirmation requests from ftp
- mget .... : downoad all files with given specification
- close: close connection
- bye: close ftp tool