One basic approach for analysis of microarray data is, to find genes / conditions which show statistical differences in their expression between different groups of conditions / genes.
To test such hypotheses you can perform parametric statistical tests (e.g. t-tests, ANOVA, Regression response), on nonparametric tests (e.g. Wilcoxon / Mann-Whitney or Kruskall-Wallis tests).
Significant genes are selected dependant on their p-value
and may be viewed as data tables, expression- / centroid-graphs or Heat maps.
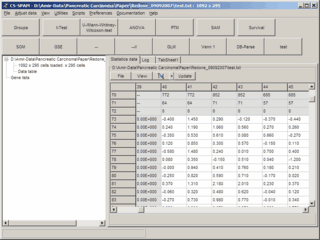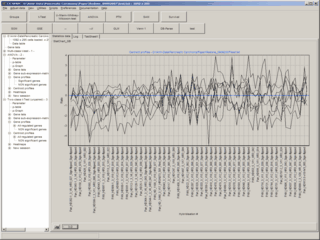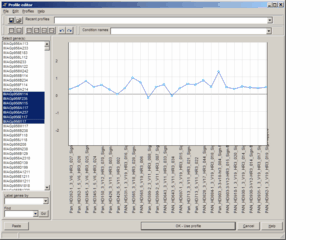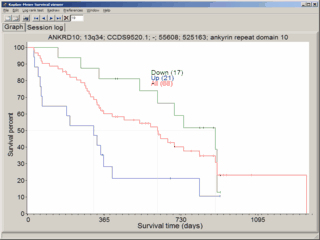
Test whether significant genesets are enriched on KEGG pathways, Gene-Ontology terms (GO), chromosomes or with miRNAs.
Sets of significant genes may be used for classification / class prediction.
Normalisation and data imputation routines allow to correct experimental
artefacts.
Diagnostic plots help to find most suited data normalisation methods.
SUMO is optimized to work with tab-delimited flat file databases.
A comfortable hybridisation selector allows to easily browse hybridisation annotation and
assign hybridisation to the analysis groups.
The whole functionality may be applied to any kind of
tab-delimited regular data files containing numerical data (e.g. miRNAs,
Proteins ... or non-biological data).
SUMO is a stand-alone MS Windows application and can be executed without installation or additional supporting libraries.