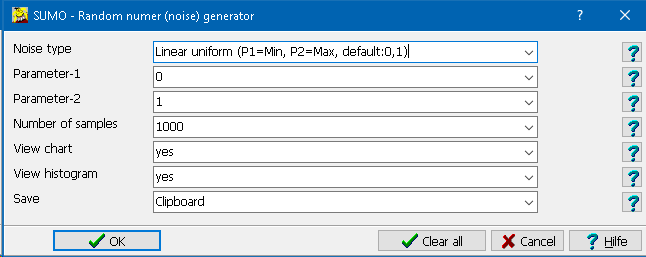
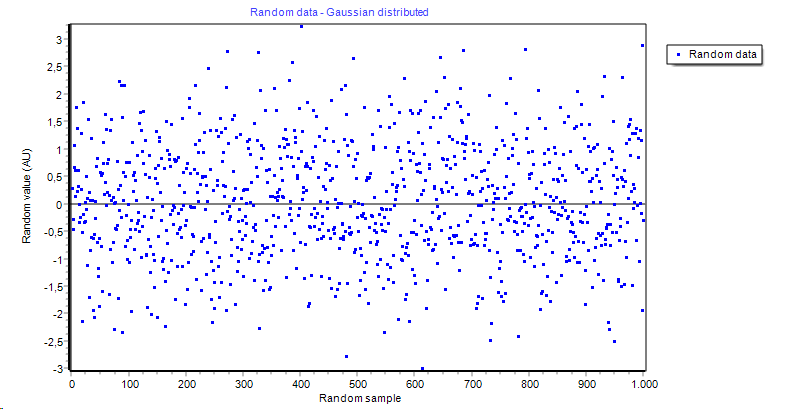
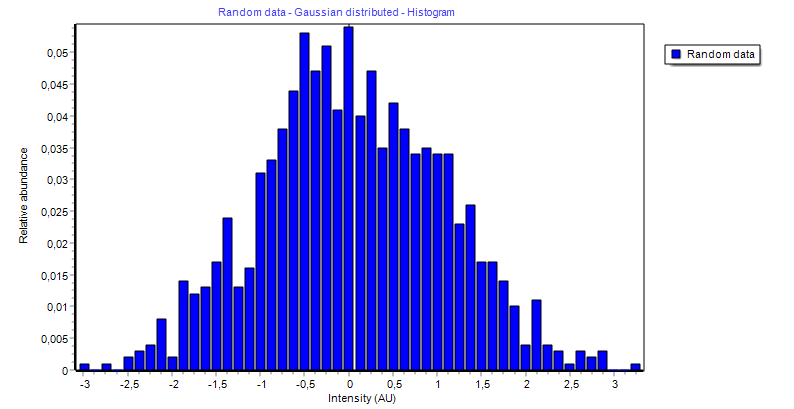
| Grubbs outlier removal | A tool to remove outliers from data vector(s) |
| Methylation | Here you can find various functions to preprocess/convert methylation data, derived from Ilumina 450K methylation microarrays. |
| library(illuminaio) idat <- readIDAT("filename.idata") write.table (idat$Quants,"filename.idat.txt",sep="\t") |
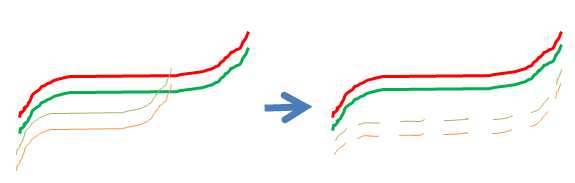

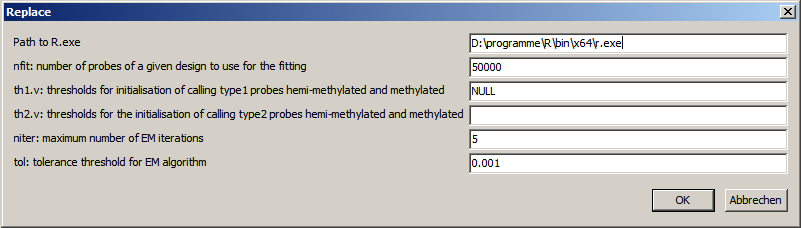
| SUMO_BMIQ_r_result.txt | Log file from R, showing any potentially problems/exceptions during execution of BMIQ R-script. |
| SUMO_BMIQ_r_input.txt | The data set exported by SUMO as nput for the R-script. The file should cntain two columns: type=Ilumna designtype (1 or 2) data=beta values (0 to 1) |
| SUMO_BMIQ_r_data.txt | Data created by the R-script, re importes by SUMO into the expression atrix. one data column containing the transformed Beta values (0 to 1) |
| SUMO_BMIQ_r_output.txt | Other computational results generated by the Rscript, here the "quality" for the iterative Beta-fits |
| SUMO_BMIQ_r_script.txt | The R-script generated by SUMO. |
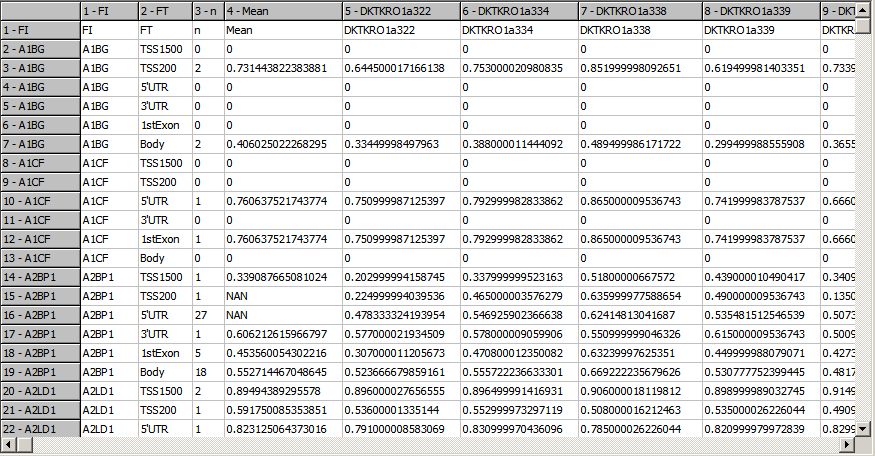
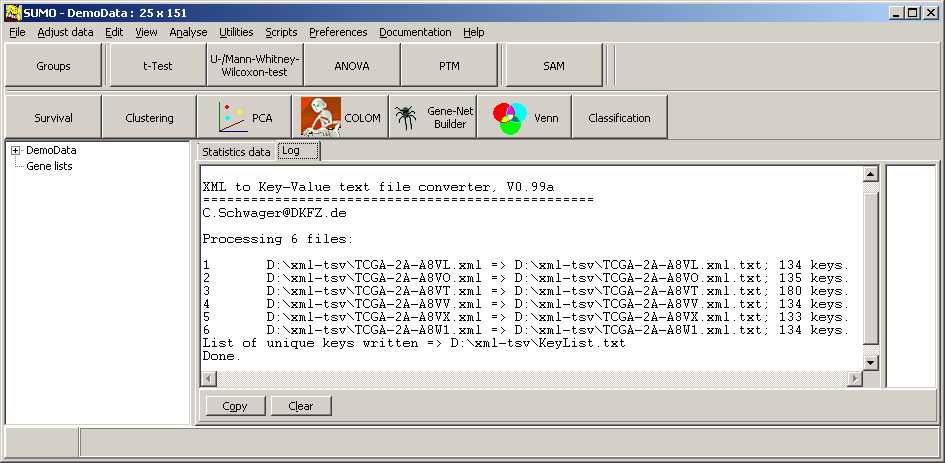
TCGA-2A-A8VL.xml -- xml -- prad:tcga_bcr -- admin:admin -- admin:bcr Nationwide Children's Hospital admin:file_uuid C2A2058A-37FA-49BE-A806-567A9A332504 admin:batch_number 389.50.0 admin:project_code TCGA admin:disease_code PRAD admin:day_of_dcc_upload 31 admin:month_of_dcc_upload 3 admin:year_of_dcc_upload 2016 admin:patient_withdrawal -- admin:withdrawn false prad:patient -- admin:additional_studies -- clin_shared:tumor_tissue_site Prostate shared:other_dx No shared:gender MALE clin_shared:vital_status Alive clin_shared:days_to_birth -18658 clin_shared:days_to_death -- clin_shared:days_to_last_followup 621 ...
{ "store": {
"book": [
{ "category": "reference",
"author": "Nigel Rees",
"title": "Sayings of the Century",
"price": 8.95
},
{ "category": "fiction",
"author": "Evelyn Waugh",
"title": "Sword of Honour",
"price": 12.99
},
{ "category": "fiction",
"author": "Herman Melville",
"title": "Moby Dick",
"isbn": "0-553-21311-3",
"price": 8.99
},
{ "category": "fiction",
"author": "J. R. R. Tolkien",
"title": "The Lord of the Rings",
"isbn": "0-395-19395-8",
"price": 22.99
}
],
"bicycle": {
"color": "red",
"price": 19.95
}
}
}
root::store:bicycle:color red root::store:bicycle:price 19.95 root::store:book:0:title Sayings of the Century root::store:book:0:author Nigel Rees root::store:book:0:category reference root::store:book:0:price 8.95 root::store:book:1:title Sword of Honour root::store:book:1:author Evelyn Waugh root::store:book:1:category fiction root::store:book:1:price 12.99 root::store:book:2:title Moby Dick root::store:book:2:isbn 0-553-21311-3 root::store:book:2:author Herman Melville root::store:book:2:category fiction root::store:book:2:price 8.99 root::store:book:3:title The Lord of the Rings root::store:book:3:isbn 0-395-19395-8 root::store:book:3:author J. R. R. Tolkien root::store:book:3:category fiction root::store:book:3:price 22.99