When you start SeedScan for the first time, all data fields are empty:
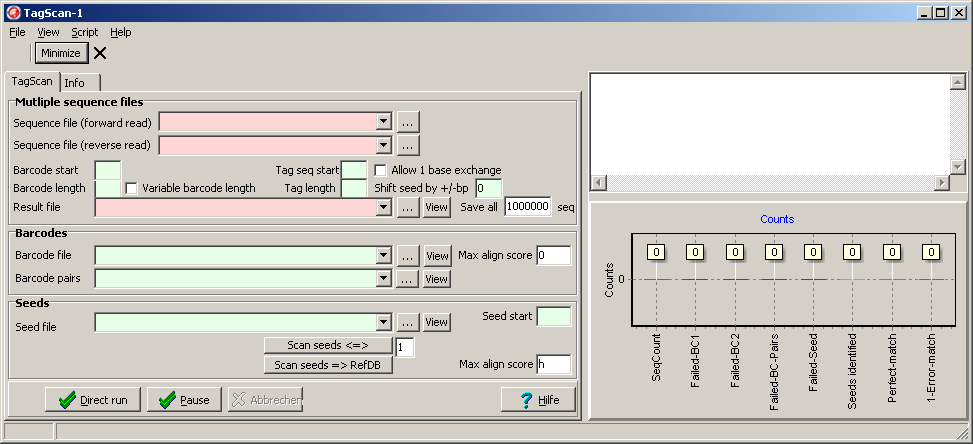
Fill in the required files and search parameters.
All red fields HAVE to be updated/specified for each analysis.
Green fields have to be updated/specified when different libraries - barcodes/pairs/seeds - are used.
White fields may be set once and remain unchanged forever.
Multiple sequence files
Define the FastQ files to be analyzed:
Drag the files from Windows File Explorer into the respective Sequence file field or
Click the "..." buttons. Use the file dialog to find and select the respective file or
Type or copy paste a file specification
FastQ files may be supplied as plain ASCII text files or as G-Zip compressed archives.
When G-Zip compressed archives are used, ensure that files have Extension "*.gz".
Next fill in structure of the constructs:
- Base position of bar code start within a sequenced construct
- Length of Bar code in bases
In case you have bar codes with different length, Check the Variable barcode length field.
SeedScan will get the individual barcode's length from the barcode list file.
- Base position of seed start within the construct.
- If required, check Allow 1 base exchange field.
This may encrease the number of identified barcodes, as a single base exchange/insertion/
deletion within a sequence against the respective barcode is tolerated.
But this may generate wrong barcode calls.
- Length of the seeds.
- If required, check Allow 1 base exchange field.
This may encrease the number of identified seeds, as a single base exchange/insertion/
deletion within a target sequence against the respective seed is tolerated.
But this may generate wrong seed calls.
- Set the Shift seed by +/- BP field to a small number (1..5) to tolerate insertions/deletions
in front of the target's seed sequence against the expected position.
This may encrease the number of identified seeds, but may generate wrong seed calls,
especially if the shift range is defined very wide (e.g. >10bp)
- Define the result file, where final seed count matrix is saved.
In case you run multiple instances of SeedScan in parallel, take care to use DIFFERENT result file names for
the different instances. Other wise the result files are overwritten.
You may save the result at regular intervals, e.g. to rescue a partial result in case the analyses gets interrupted.
Define the number of sequence pairs after which the result file is created/updated automatically.
E.g. Define "10000000" to save the result after any 10 Mega sequences have been analyzed.
Don't save to often: saving, especially on network mounted drives, may consume a mentionable amount of time.
- Click the View button to see the present result matrix.
Bar codes
Supply:
- Barcode file: A tab delimited text file containing a list of barcode sequences used for this analysis
Best would be to only specify barcodes used in this analysis.
Additonal barcodes - not used for multipexing in the particular analysis - will not generate problems but waste processing time.
For details about file format => see Data files.
- To preview the selected barcode/pairs list click the View button.
- Barcode pairs: A tab delimited text file containing a list of bar codes pairs (and optional target library index).
For details about file format => see Data files.
Seed files
- Seed file: A tab delimited text file containing a list of seed sequences used for this analysis.
You may drag multiple seed sequence files into this field, one after the other.
SeedScan will scan each barcode-pair's target sequence agianst the respective library as defined in the pairs list.
Best would be to only specify seeds used in ths analysis.
Additonal seed sequences - not used in the particular analysis - might generate erraneous seed calls and waste processing time.
For details about file format => see Data files.
To preview the selected barcode list click the View button.
- Seed start: The position of the seed sequence within the seed list files sequences.
Run the analyses
Now, after all parameters have been checked and correctly adjusted, you may start the analysis.
Click the Direct Run button.
The Library files are loaded.
Loading and contstruction of 64-bit sorted hashcode transformed seed libraries will take a while.
Transformation progress is indicated in the status line (bottom of window).
Next scan is started.
Scan progress is indicated by regular update of the bar chart and in the program windows title line.
You may click the Minimize button to reduce SeedScan's window but still see the scan progress:

We can see:
- A total of 289.7 Million reads were already scanned.
- 176.7 Million seeds <=> target have been identified
- Average analysis speed is ~ 51.000 analyzed sequences/secod
- Total elapsed analysis time of ~94:15 min
At any time, you may:
- View any of the diagnostic plots
For details see Diagnostic plots.
- Pause the process by clicking the Pause button. E.g. to view an intermediate result matrix
Resume a paused process by clicking the Resume button.
- Cancel the ongoing analyses by clicking the Cancel button.
This will stop the analysis and save the partial data generated data until the cancel-timepoint.
The analysis finalizes as soon as:
- all data from the FastQ files have been read and scanned.
- Canceled by user
ClonTracer - Run the analyses
For ClonTracer runs, remember the expected structure:
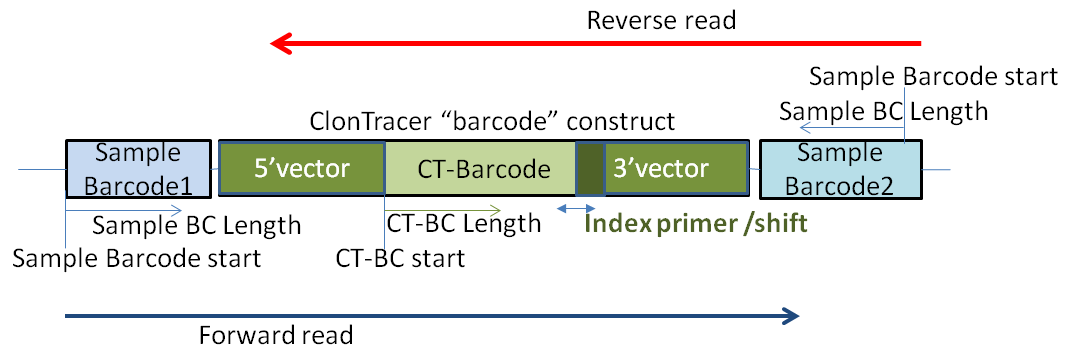
Now, after parameters have been checked and correctly adjusted:
- Sequnece files, Forward/reverse
- Barcode- / Pairs-file
- Barcode Start/Length/Variable barcode length
- Taq seq start => CT-BC start
- Tag lenth => CT-BC length
-
- Index primer => The last few bases of the ClonTracer 3'vector sequence
(i.e. the next few bases following the ClonTracer barcoe. - >
- Index primer shift (+/- BP range): search range for the index primer;
compensating for false inserted/deleted bases by sequencing.
- Result file
you may start the analysis.
Click the ClonTracer Run button.
The Library files are loaded.
Next scan is started.
Scan progress is indicated by regular update of the bar chart and in the program windows title line.
You may click the Minimize button to reduce SeedScan's window but still see the scan progress:
At any time, you may:
- View any of the diagnostic plots
For details see Diagnostic plots.
- Pause the process by clicking the Pause button. E.g. to view an intermediate result matrix
Resume a paused process by clicking the Resume button.
- Cancel the ongoing analyses by clicking the Cancel button.
This will stop the analysis and save the partial data generated data until the cancel-timepoint.
The analysis finalizes as soon as:
- all data from the FastQ files have been read and scanned.
- Canceled by user
For each sample a ClonTracer Barcode list is generated, containing all CT-BarCcodes/abundance for this specific samples.
- A tab-delimted text file
- one CT-Barcode per line
- tab-delimited:
- Line count
- Barcode - numeric representation
- Barcode sequence
- Abundance of this particular CT-BArcode
Additionally a consensus matrix will be generated.
By default, all CT-Barcodes which show up in at least one sample with abundace>=100 will be collected into the consensus matrix.
Use ClonTracer Utilities to:
- Filter potential error CT-Barcode sequences
- Create a custom filtered consensu matrix
- Generate Base-Composition logos
- Generate histograms
- Copy sequences from CT-BC list for e.g. Venn analyses.
Result data
Finally, as soon as the anaylsis has terminated, a set of result files are automatically generated:
- Result matrix: A tab delimited table containing count numbers for all samples/seeds specfied in the analysis.
For details about file format => see Data files
.
("basefilename_Matrix.txt")
- Summary: A text file summarizing process parameters and global counts (~values shown in the bar chart)
("basefilename_Summary.txt")
- Failed-BC1: A text file with the first ~1000 sequence pairs where no valid Barcode-1 was detected.
May be useful for error analysis
("basefilename_Failed_BC1.txt")
- Failed-BC2: A text file with the first ~1000 sequence pairs where a valid Barcode-1 was detected,
but no listed Barcode-2
May be useful for error analysis
("basefilename_Failed_BC2.txt")
- Failed-BCPairs: A text file with the first ~1000 sequence pairs where Barcode-1 as well as Barcode-2
were within the defined list, but pairing is not "allowed".
May be useful for error analysis
("basefilename_Failed_BCPairs.txt")
- Failed-IndexPrimer: A text file with the first ~1000 sequence pairs where IndexPRimer was not found.
- Short Report collecting process parameters and diagnostic plots.
("basefilename_Report.htm")
All result files are generated in the location as specified for the Result file.
The differerent result file names are composed of the base file name - as defined by the user - and an extension specific for the data type:
View menu
From the view menu you may ona any of the disbostic plats:
Utilties menu
From the utilities menu you may select extended options or additionaL functionality not diretly
related to seed scanning
Rescue no-BC1 sequences
Due to the implementation of base calling software on Illumina HTS systems, highly similar costructs
may result in a mentionable fraction of miscalled sequences.
To circumvent this problem, it may be helpful ta add 20%-50% of random non-sense sequences to the sequencing library.
This should increase the number of correctly called sequences - but on the other hand will reduce the fraction of useable sequences.
In the special case of our barcoded seeds library constructs, the first ~35 bases are mostly common (~30 different version),
followed by ~60000 different seeds.
Under this condition even a Transrciptome/Exome/Genome/Methylome/ ..... library might act as "non-sense" library.
Thus you might use the capacity of the "non-sense sequences" (~100 Million reads on a HiSeq4000) to run additonal meaningful samples.
Check the Rescue no-BC1 sequences option.
All Sequence pairs where no valid Barcode-1 was found will be written into 2 new FastQ files.
These files may be used subsequently for alignment to a genome or whatever else.
The new FastQ files will be created with names:
- "Base_Result_Name_Rescued1.fastq" for the forward reads
- "Base_Result_Name_Rescued2.fastq" for the reverse reads
Writing this files will slow down the analysis progress and create big files (~200 GByte).
Thus, acitvate this option only if you really want to reuse the "non-sense" sequences.
Scan seeds <=> Seed-Lib
Accuracy of seed identification depends among sequencing quality on uniqueness of the seed sequences.
To check this, you may scan each individual seed sequence against the complete seed library.
In an ideal case you would only find unique matches
Even if 1-error searching is enabled.
One seed file may be checked in a single analysis.
The seed file is taken from the Seed file text field. If two (or more) files are specified, the first one is used.
Check the Allow 1 base exhcange box to enable 1-error matches.
SeedScan generates a brief summary and two result files:
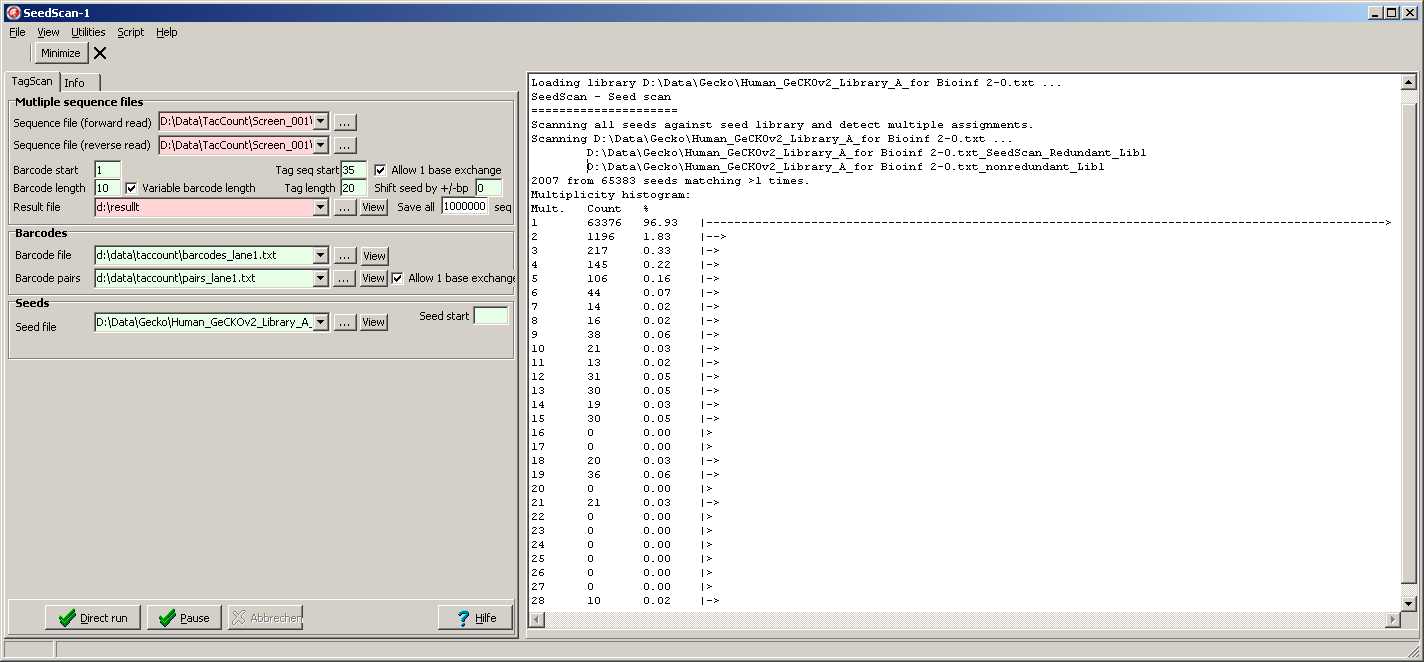
The histogram indicates the distribution of sizes from crossmatching clusters.
As exprected far most of the seeds are singular.
But there are also 10 cluster where 28 seed's targets share the same recognition sequence.
Two data files are generated:
- "*_nonredundant_Lib1" - a list of unique seeds.
- *_Redundant_Lib1 - a list of clusters, each cluster containg a set of seeds sharing the same recognition sequence:
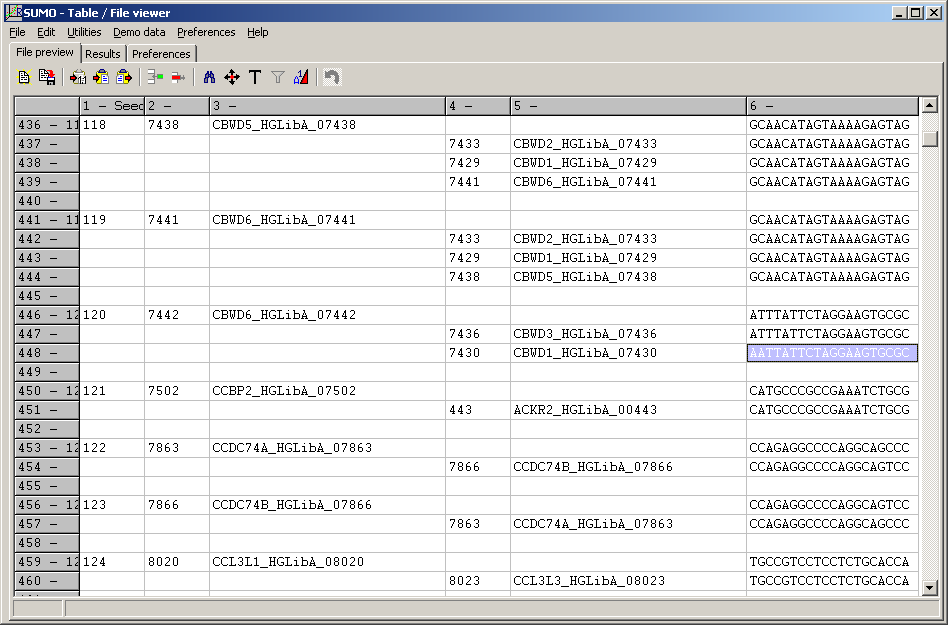
- First three columns show Number, ID and Name of intial seed, building a cluster.
- Colmn 4 and 5 show ID and name of the other seeds with identical sequence
- Column 6 shows the seeds' sequneces.
- Clusters are separated by one empty line
- In the example, cluster 120 contains also a 1-error matching seed:
CBWD1... / CBWD6 - AATTATTCTAGGAAGTCGC / ATTTATTCTAGGAAGTCGC
Scan seeds <=> Reference library
Another question arises:
- do the seed sequences match uniquely the desired sequence target or
- multiple sequencer or
- none at all ?
Therefore, you may scan all seeds against a reference sequence library and find out (for each individual seed sequence:
- do I get a unique match on the refernece library (e.g. seed sequeuce => to a single RefSeq gene)
- is the name of the seed sequence the same as the reference's name (or may be a gene alias name)
- do i get multiple matches, what are the reference's gene names
Example:
You have a libray of gene silencing constructs for human genes.
Each construct contains a guide sequence (the seed sequence) which shall target a single gene.
Donwload a list of all genes as multiple FASTA file - e.g. RefseqRNA for homo sapiens from NCBI's ftp server:
ftp://ftp.ncbi.nlm.nih.gov/genomes/H_sapiens/RNA/
Download and uncompress "rna.fa.gz".
Process your seed library that each line contains tab delimited:
- Gene symbol
- seed sequence
E.g.:
Seed_name Seed_seq
A1BG GTCGCTGAGCTCCGATTCGA
A1BG ACCTGTAGTTGCCGGCGTGC
A1BG CGTCAGCGTCACATTGGCCA
A1CF CGCGCACTGGTCCAGCGCAC
A1CF CCAAGCTATATCCTGTGCGC
A1CF AAGTTGCTTGATTGCATTCT
A2M CGCTTCTTAAATTCTTGGGT
A2M TCACAGCGAAGGCGACACAG
A2M CAAACTCCTTCATCCAAGTC
A2ML1 AAATTTCCCCTCCGTTCAGA
...
Select Main menu | Utilities | Seeds => RefDB.
A Parameter dialog opens up:
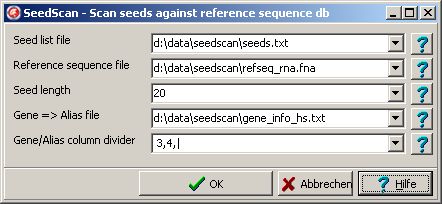
Fill in required values for seed-list / reference-db files and seed length.
Double click the respective fields to open a file selection dialog
or more easily just drag files from Windows Explorer into the respective fields.
To check gene alias names too, supply a file mapping "official" gene symbls to commonly used gene aliases in the Gene => Alias file field.
Such a file may be downloaded from NCBI's ftp server.
E.g. "Homo_sapines_gene_info":
#tax_id GeneID Symbol LocusTag Synonyms dbXrefs chromosome map_location description type_of_gene Symbol_from_nomenclature_authority Full_name_from_nomenclature_authority Nomenclature_status Other_designations Modification_date Feature_type
9606 1 A1BG - A1B|ABG|GAB|HYST2477 MIM:138670|HGNC:HGNC:5|... 19 19q13.43 alpha-1-B glycoprotein protein-coding A1BG alpha-1-B glycoprotein O alpha-1B-glycoprotein|HEL-S-163pA|epididymis secretory sperm binding protein Li 163pA 20170903 -
9606 2 A2M - A2MD|CPAMD5|FWP007|S863-7 MIM:103950|HGNC:HGNC:7|... 12 12p13.31 alpha-2-macroglobulin protein-coding A2M alpha-2-macroglobulin O alpha-2-macroglobulin|C3 and PZP-like alpha-2-macroglobulin domain-containing protein 5|alpha-2-M 20170903 -
9606 3 A2MP1 - A2MP HGNC:HGNC:8|Ensembl:ENSG00000256069 12 12p13.31 alpha-2-macroglobulin pseudogene 1 pseudo A2MP1 alpha-2-macroglobulin pseudogene 1 O pregnancy-zone protein pseudogene 20170402 -
9606 9 NAT1 - AAC1|MNAT|NAT-1|NATI MIM:108345|HGNC:HGNC:7645|... 8 8p22 N-acetyltransferase 1 protein-coding NAT1 N-acetyltransferase 1 O arylamine N-acetyltransferase 1|N-acetyltransferase 1 (arylamine N-acetyltransferase)|N-acetyltransferase type 1|arylamide acetylase 1|monomorphic arylamine N-acetyltransferase 20170906 -
9606 10 NAT2 - AAC2|NAT-2|PNAT MIM:612182|HGNC:HGNC:7646|... 8 8p22 N-acetyltransferase 2 protein-coding NAT2 N-acetyltransferase 2 O arylamine N-acetyltransferase 2|N-acetyltransferase 2 (arylamine N-acetyltransferase)|N-acetyltransferase type 2|arylamide acetylase 2 20170903 -
The tab-delimited file should contain:
- one gene per line
- one column containing the gene symbol
- one column containing muliple aliases/synomymes divided by a special divide character
In the Gene/Alias column divider field define (delimited by comma (","));
- ID of column containing the gene Symbol.
In the example column 3
- ID of column contaning the aliases/synonyms.
In the example column 5
- Divider
In the example pipe ("|")
Click OK-button to start the scan.
Each individual reference sequence is scanned aginst the seeds, accepting
- perfect matches
- forward as well as reverse (reverse/complement) sequence
For each seed the number of matching reference is counted and the names between the seeds and the reference library are compared.
Several result files are generated:
" *_Ref_UniqueMatch - the list of genes which match their target unqiuely on a reference sequence which has the same "name" (or an alias) as the seed.
" *_Ref_MultipleMatches" - a list of seeds which match multiple reference sequences.
Each line contains (tab delimited) Seed name, Seed sequence, Seed-ID, List of names from matching sequences
E.g.:
SeedName SeedSeq SeedID Matched gene symbols
ANKRD65 CACCCGCGAGTGTCCGTGCC 2137 ankrd65,loc105378585,
ANKRD65 CCGCCTGGCACGGACACTCG 2138 ankrd65,loc105378585,
APITD1 GGCAGCAGTTCACTATACTG 2424 apitd1,apitd1-cort,
APITD1 GAGCTGACTTTCCGACAGTG 2425 apitd1,apitd1-cort,
APITD1-CORT GCAGCGATTCTCTTACCAAC 2427 apitd1,apitd1-cort,
APITD1-CORT GGCAGCAGTTCACTATACTG 2428 apitd1,apitd1-cort,
APITD1-CORT GAGCTGACTTTCCGACAGTG 2429 apitd1,apitd1-cort,
ATAD3B GTACAGGCCCCGGTTCTTCT 3413 atad3b,atad3c,
BAI2 GCGCCGCTTCCGCATGTGCC 4115 adgrb2,bai2,
BAI2 CGAAGTTCTTGTCGAAGTGC 4116 adgrb2,bai2,
BAI2 CTACCTGCGCTTCAACCGCC 4117 adgrb2,bai2,
BMP8B GGTCATGAGCTTCGTTAACA 4623 bmp8b,loc105378951,
C1orf151-NBL1 TAGCCCGATTCATGCCATCT 5776 minos1-nbl1,nbl1,
C1orf151-NBL1 CGGCAAGGAGCCTAGTCACG 5777 minos1-nbl1,nbl1,
C1orf151-NBL1 TCGCACCAGGCACTCTTATC 5778 minos1-nbl1,nbl1,
CCDC28B TGAGGAGCAGTCCGCTGCGT 7747 ccdc28b,tmem234,
CDK11A AAGAAAGAGAGCACGAACGT 8724 cdk11a,cdk11b,
CDK11A AAGAACAGGATAAAGCTCGC 8725 cdk11a,cdk11b,
CDK11A AACCACCCCAGCAAATGTCT 8726 cdk11a,cdk11b,
CDK11B TGCCTCCTCAGAGTTGCTGC 8728 cdk11a,cdk11b,
CDK11B ACATCACCGAACGATGAGAG 8729 cdk11a,cdk11b,
CLCNKB GAAGACCATGTTGGCGGGTG 9843 clcnka,clcnkb,
CORT TGTCCCGGCGCGCAGATTGC 10796 apitd1-cort,cort,
CORT CCCCCCAGCAATCTGCGCGC 10798 apitd1-cort,cort,
DCDC2B TTAATTCGGTCTGGTTCCTT 12487 dcdc2b,tmem234,
DCDC2B GGTAACCCAGCCCTCTCCAA 12488 dcdc2b,tmem234,
EPHA2 CTACAATGTGCGCCGCACCG 16289 epha2,loc101927479,
GJA9 GAGCCGCTATTTAAGTGCCA 20153 gja9,gja9-mycbp,
GJA9 CAGAAATGTATGCTACGACC 20154 gja9,gja9-mycbp,
GJA9 CCAATCGCAGTTAGCTGGCA 20155 gja9,gja9-mycbp,
GTF2H2D CACCTGTAATCCCAGCTACT 21553 c1orf86,cep104,cldn19,draxin,fam110d,hp1bp3,loc105376829,nol9,nudc,otud3,pik3cd,sepn1,slc25a34,smim12,tnfrsf9,zbtb8a,zmym1,
HMGB4 ACTCGACAAAGCCCGATACC 22698 c1orf94,hmgb4,
HNRNPCL1 CACTCTTGTTGTCAAGAAAT 22811 hnrnpcl1,hnrnpcl2,hnrnpcl3,hnrnpcl4,
hsa-mir-1254-1 GTGCCACTGTACTCCAGCCT 23466 loc105376896,pqlc2,psmb2,
hsa-mir-1273a TCGCCCAGGCTGGAGTGCAG 23585 akr7l,bmp8a,c1orf109,dffa,fbxo44,loc105376835,loc105378653,man1c1,maneal,meaf6,nfyc-as1,slfnl1-as1,snip1,tnfrsf1b,zbtb8a,
hsa-mir-1273a GCGCCACTGCACTCCAGCCT 23586 bmp8a,bmp8b,c1orf109,cdc42,cep104,cfap74,dffa,ece1,fbxo44,h6pd,hes2,loc101928303,loc101928391,loc105376858,loc105378666,nfyc,nol9,pafah2,phactr4,rab42,rhbdl2,rnf19b,slc35e2,slfnl1-as1,smim12,smpdl3b,
hsa-mir-1273d TCACCCAGGCTGAAGTGCAG 23595 loc105376864,mir1273d,
hsa-mir-1302-5 CAGGCATGAGCCACTGTGCC 23763 htr1d,nipal3,snip1,
hsa-mir-200b TCTTACTGGGCAGCATTGGA 24208 mir200a,mir200b,mir429,
...
" *_Ref_WrongMatch" - list of genes with a unique perfect match to a reference sequence, but different sequence names.
SeedName SeedSeq SeedID Matched gene Symbol
ABP1 GATCCAGCGCTGGACGTAGT 319 aoc1,
ACPL2 GAAACCGTATCACCCAAAAC 520 pxylp1,
ADC GTACACGATGGTCTTGGAGG 886 azin2,
ADORA3 TCCGCAAGGCTGACCGCTCC 1018 tmigd3,
AGPHD1 ATCATGTTTCTGAAAGCCGC 1237 hykk,
AGXT2L1 CAACGACTTAGCCTTACGCC 1276 etnppl,
AGXT2L2 GCAGTACATGTACGATGAAC 1279 phykpl,
ALS2CR8 TTGACTCTTCACCGTCATTA 1729 carf,
ANAPC15 CCCACAGCTCCAAAGCATCG 1875 lrtomt,
ANLN AACTCACTCACCTCCGTAAA 2168 KIAA0895,
AQPEP GTTCCAGCTGGGACGCTAAC 2613 lvrn,
ATP4B TCTCTCCAGGGGTAACCTTA 3636 znf510,
ATPBD4 CTCTATCGCCGAACCATAAG 3803 dph6,
AZI1 TCACCTTGCCATCCAATGCC 3933 cep131,
B3GNT1 CTGCCGAAAGCGCTCGTCGA 3980 B4GAT1,
BCMO1 GTCGAACCAATGGTTGTATC 4355 bco1,
BET3L TAACTCAGCTAGCCGCCCCG 4437 trappc3l,
C10orf114 AATCGCTAGCTGCGCATCCG 5036 CASC10,
C10orf118 GCAGAACATCGAGTACCAAA 5039 ccdc186,
C10orf129 AAGTTCCCAAGACAGCGCTC 5053 acsm6,
C10orf137 ACTCTTTATGAGATCGTCTC 5057 edrf1,
...
" *_Ref_NoMatch" - list of genes where theres is no perfect match of the seed sequnece to any sequence in the reference library.
But there may well be incomplete, shortened or error containing matches.
Random data
For demonstration or test purposes you may generate a complete random dataset.
From main meu select: Main menu | Utilities | Random data.
A parameter dialog opens up:
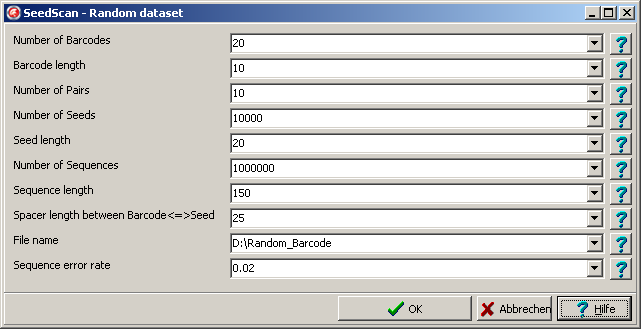
Set all parameters accordingly.
The File name is appended with respective extension for the different filetypes.
E.g. Barcode file: "Base_Name_Barcodes.txt", ....
Preview FastQ
To verify positions of barcodes/seeds within the FastQ sequences, it may be helpfult to preview a FastQ file.
What sounds trivial may become complicated with Giga-Byte files or GZ-compressed archives.
Select Main menu Utilities | Preview FastQ.
Select a plain text FastQ file or a GZ-compressed file (assure GZ-files have file extension (".GZ").
The first thousand lines from the selected file are loaded and shown in a generic table viewer.
Instead of FastQs you may preview any kind of huge (GZ-compresed) text files.
FastQ => FastA
You might want to anlysze FastQ files with tools which only accept FastA files.
This utility converts FastQ => FastA Format.
It simply removes the Direction and Quality line and converts the sequence start sign "@" into the standard ">" sign.
Select Main menu | Utilities | FastQ=0> FastA.
Select one - or multiple - FastQ files (either plain text or GZ-copressed [ with extension ".gz"]).
The new files will be created with extension .fasta.
Split FastQ
This options allows to split a FastQ file using the Sample BarCode pairs.
Define in the main SeedScan tab:
- Sequence file (forward read)
- Sequence file (reverse read) - optional
- Smaple Barcode file
- Sample Barcode pairs - optional)
- Sample Barcode start
- Sample Barcode length
- Variable Sample Barcode length
- Allow 1 base exchange (in Sample Barcode)
Select Main menu | Utilities | Split Fastq
For each sample barcode pair - as defined in sample barcode and pairs file - individual FastQ files for forward and reverse sequences are created.
(Originalname_S01.FastQ ... OriginlaName_SXX.FastQ).
SeedScan analyzes the input files:
- Sequences without BC1/BC2 or invalid pairs are skipped
- Matching sequences (BC1 AND BC2 AND valid pairs) are written to the respective new FastQ file
Reading and Writing of multiple files is time consuming.
To avoid excessive disk access, SeedScan buffers output and writes result file in 10MByte junks.
Nevertheless, spliting a FastQ file into 30 samples (i.e. 30 barcode pairs, forward and reverse reads, 250bp) runs at ~25 Kilo-Sequences/s on a local spinning disk.
Running the same task accross a network mounted disk (1-GigaBit network) reduces the processing speed to ~1 Kilo sequences/s.
ClonTracer utilities
Here you may find some options to filter / analyse ClonTracer List files.
Agglomerate error barcodes
During a ClonTracer run a library of ClonTracer Barcodes is extracted from the analyzed sequences.
The library will contain:
- all unique ClonTracer Barcodes
- variants from ClonTracer barcodes created by sequencing errors:
- Base exchanges
- Insertions
- Deletions
Obviously, there will be:
- a high probability for error sequences from highly abundant Barcodes
- a high probability that very low abundant Barcodes are in reality error variants from the highly abundant ones.
At an expected sequencing error rate from 1%-5% we could expect 0.3 to even 2 sequencing errors within a 30 BP ClonTracer Barcode.
Thus we could apply a filtering strategy:
- Use all high abundant Barcodes, e.g. those with count ≥1000.
-
- Compare all low abundant Barcodes, e.g. those with count ≤10.
- If such a low abundant barcode is a variant of a high abundant Barcode,
just remove it from the list and add its counts to the one of the mother Barcode.
- Use base exchange variants (1 or 2)
- Use Insertion / Deletion variants (1 or 2 bases),
eventually even with base exchange variants on the Insertion/Deletion vartiants.
Filtering a ClonTracer barcode list with 10 million sequences and 1 be/ins/del requires less than a secod.
Filtering allowing 2 errors (2xbe /2xins / 2xdel / ins+be / del+be / ...) may require minutes.
Select SeedScan Main menu | Utilities | CloncTracer Utilities | Agglomerate error barcodes.
In the file selection dialog, select 1 or multiple ClonTracer Barcode lists.
A parameter dialog opens up:
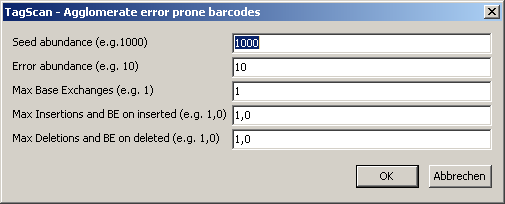
Define the parameters:
| Seed abundance |
Minimum number of counts for the high abundant barcodes |
| Error abundance |
Maximumn number of counts for the potential error barcodes |
| Max base exchanges |
Max Number of allowed base exhcange errors |
| Max insertions and BE on inserted |
Max Number of allowed insertions,
and optional number of base exchange errors on the "inserted" barcodes |
| Max deletions and BE on inserted |
Max Number of allowed deletions,
and optional number of base exchange errors on the "deleted" barcodes |
It may be a wise strategy to:
- First round, capture the simple errors (1 exchange/insertion/deletion),
at high seed and low error abundance
- in next rounds
- lower seed abundance, increase error abundance, or
- increase number of allowed errors (base exchange/insertion/deletion), or
- allow additional base exchanges on insertions/deletions.
E.g. define Max Insertions = "1,1" to generate all inserton variants (~120 for a 30 BP barcode),
and on each single insertion variant, all 1 base exchange variants (~120*120 = 14400 variants).
Filtered CT-Barcode lists are created as new files like "OldFileName_new".
Results and statistics are shown in SeedScan's log window:
SeedScan - ClonTracer utilities - Agglomerate error sequences
====================================================================
Ver. 1.001a from 09.07.2018, C.Schwager@DKFZ.DE
Parsing 1 data files ...
# Name #-TC-BC #-Seq
1 U:\User\Project AN4-ClonTrac\HiSeq4000_11120\180607_ST-K00207_0156_BHVLVMBBXX\AS-243465-LR-35510\fastq\Result_CTCounts_1.txt 650509 14116053
Filtering parameters:
Seed abundance: 1000
Error abundance: 10
Max Base Exchanges: 1
Max Insertions and BE on inserted: 1
Max Deletions and BE on deleted: 0
Processing file 1: U:\User\Project AN4-ClonTrac\HiSeq4000_11120\180607_ST-K00207_0156_BHVLVMBBXX\AS-243465-LR-35510\fastq\Result_CTCounts_1.txt
# of Base Exchanges: 1
#Seeds: 369 # Merged BC: 12252 # Merged Seq 39144 ET: 0.031s
# of Insertions: 1 and # of BE ont the inserted: 0
#Seeds: 333 # Merged BC: 2483 # Merged Seq 8528 ET: 0.031s
#Total Seeds: 702 # Total Merged BC: 14735 # Total Merged Seq 47672 ET: 0.078s
REsultfile: U:\User\Project AN4-ClonTrac\HiSeq4000_11120\180607_ST-K00207_0156_BHVLVMBBXX\AS-243465-LR-35510\fastq\Result_CTCounts_1.txt_new
from 650509 CTBC,14116053 Seqs to 635774 CTBC,14116053 Seqs; 2.27%
Done.
Additionally, the Log is saved to a file "FirstListFileName_Agg_Err_Log.txt" in the folder where the list files are found.
Filter WSWS
ClonTracer barcodes are synthesized in such a way that hybridized ClonTracer-Target complexes forms an alternating series of
- basepair interconnected by three hydrogen bonds = Strong = S (C=G)
- basepair interconnected by two hydrogen bonds = Weak = W (A=T)
Consequnently, extracted ClonTacer barcodes MUST reflect this pattern.
Extracted barcodes not showing the WSWSWSWS pattern are either synthesis or cloning artifacts or most probably sequening errors.
It may be recommended to remove such barcodes, especially if multiple errors occur.
Build consensus matrix
This option allows to interactively build a filtered consensus matrix from all selected ClonTracer barcode lists.
You may define filters for:
- Minimum number of barcode counts found at lest on sample for the respective barcode
- Minimum number of samples with count > 0 for the respective barcode
Barcodes passing both criteria are added to the consensus matrix.
Samples which do not contain the particular CT-BC will get "0" for the respective data cell,
all others will get the respective samples/barcodes count number.
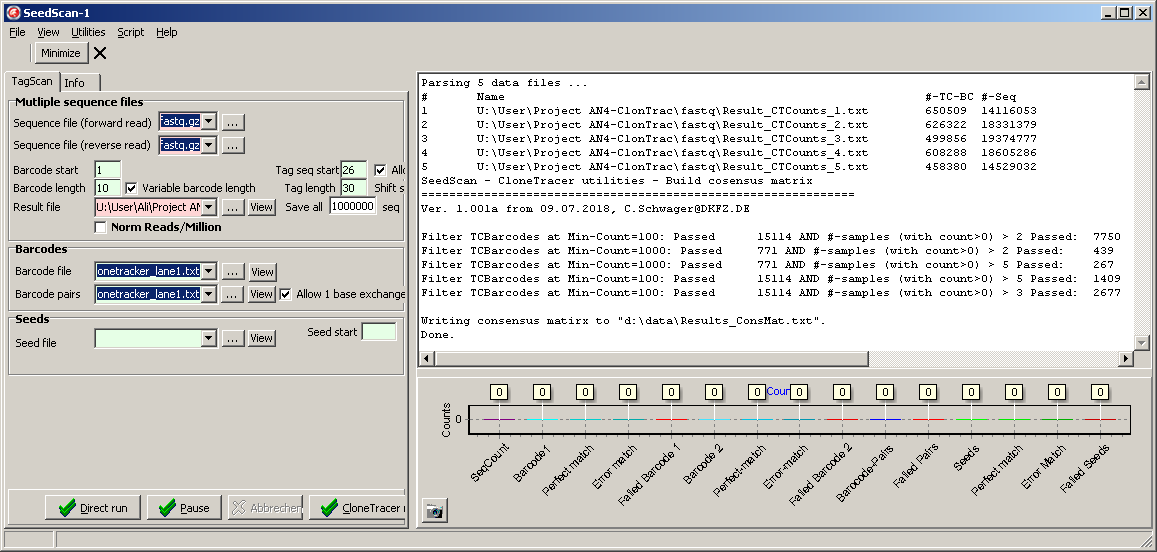
Select SeedScan main menu | Utilities | ClonTracer Utilities | Build consensus matrix.
Select at lest two ClonTracer barcode list files you want to merge into a consensus matrix.
A parameter dialog opens up:
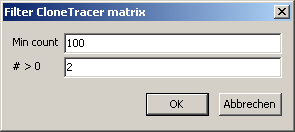
Adjust the parameters:
| Min count |
The minimum count number found in at least one of the selected sample for the respective barcodes |
| # > 0 | Minimum number of samples with count > 0 for the respective barcode |
As higher these filter parameters, as lower the nubmer of passed barcodes,
but, also, as higher the probability the filtered are useful for subsequent analysis and not only data noise.
Click the OK-button.
SeedScan counts the passing ClonTracer barcodes and shows the result in the Message log:
Now you may modify the filter parameters to see the results for higer / less stringent filtering parameters.
As soons as you are satisfied with filter settings, click the Cancel-button.
Now the matrix is build and saved, applying the last selected filters.
Base composition models
Is there a difference in the ClonTracer barcode library between differently handled/treated samples?
One way to visualize the overall composition could be to visualize position dependant base-composition as a "Sequence logo":
- for each base position a stack of 4 base characters is shown
- the total hight of a stack indicates the conservation of a base.
If only one base shows up at a position => the stack is highest,
If all 4 bases show up at same proportion => stack is lowest.
- Width of a stack indicates proportion of bases vs. ambiguites.
If all sequences contain a base (A,C,G,T) at the respective position => stack is widest.
As more ambiguities (or gaps, missing bases) as smaller the stack
- Within a stack, height of a base indicates its relative proportion at this sequence position.
- Bases are sorted by their relative frequency: most frequent top,... least frequent bottom.
Sequence logos are directly extracted from ClonTracer barcode lists (containg hundred thousands of barcode sequences).
But you may also use other file types:
- Tab delimited text files:
- first (header) line ignored
- one sequnece per line
- each line tab delimited
- i-th tab contains a (DNA) sequence
e.g. the seed files (gecko library)
- Multiple FASTA files
Sequence logos for muplitple samples are generated and collected in an html file.
For each barcode list two sequence logos are generated:
| CT-BC logo |
is computed from all identified unique barcodes
|
| Abundance count weighted logo |
is computed by weighting each individual barode by it's abundance.
Thus highly abundant barcodes may alter the overall base compostion sequence model.
|
Select SeedScan main menu | Utilities | ClonTracer Utilities | Base composition.
Select one or mutiple ClonTracer barcode list you want to analyze.
The resulting html file contains:
- Relativ base composition tables
- Sequence logos
for both methods:
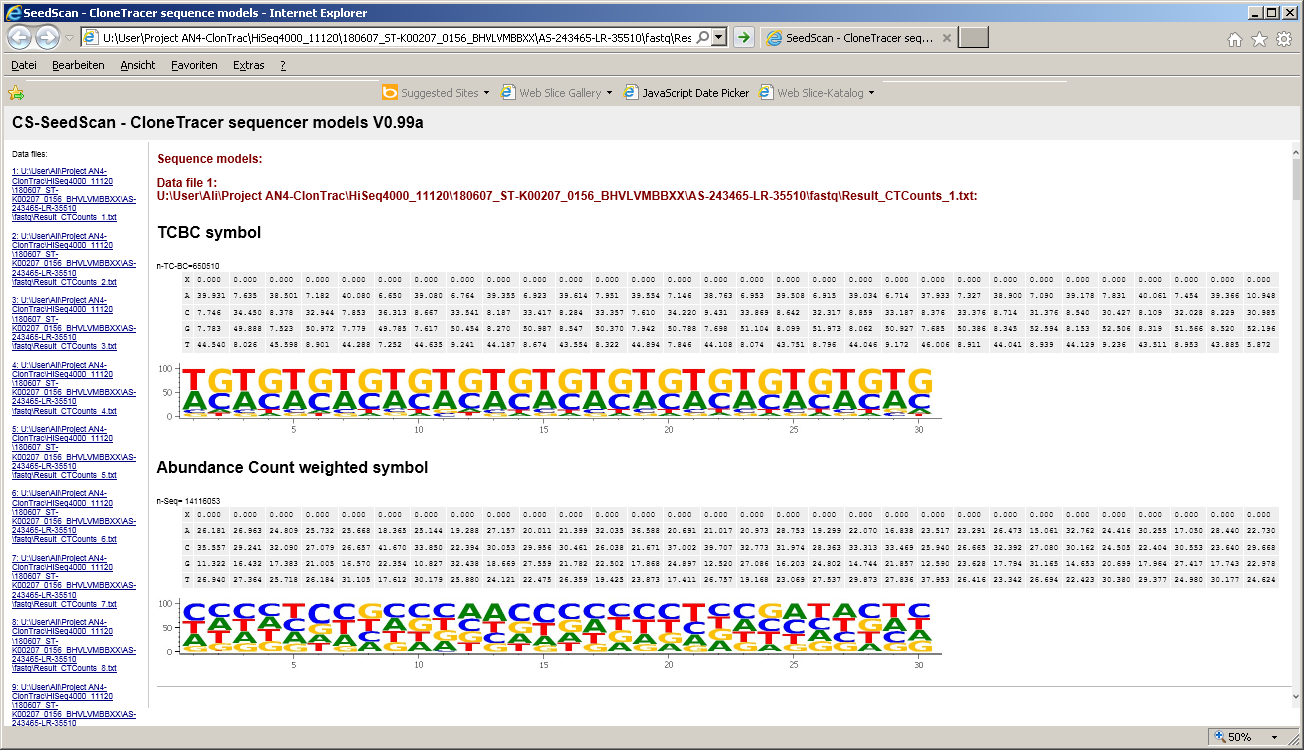
And there may be substantial differences between samples and methods.
Merge ClonTracer matrices
This tool may be used to merge consensus matrices build with SeedScan (see above).
The problem here is: result matrices from different ClontTracer analyses may contain differint BC-sequneces.
Thus, matrices can not be appended row wise, but they must be sorted to combine common CT-BCs, or add "spcers" for unique CT_BCs coming from the individual matrices.
Although designed for ClonTracer result matrices, But you may merge any kind of isomorphic data matrices.
SeedScan expects tab delimitd text files with:
- Same number of header rows
- Same number of colums rows
- Identical key column (here ClonTracer bar-code sequence column), with unique keys
- First header row, contains names for data columns
- Data cells should contain numbers
Non numeric cells are converted to "0" for computation of row/colun sums
Select SeddScan | Main menu | Utilities | ClonTracer | Merge tables|.
A parameter dialog opens up:
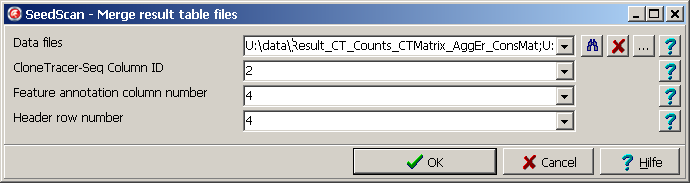
- Drag (multiple) data files from Windows explorer into the Data files field.
Repeated Drag/Drop operatons will just append the new files to the list of selected.
Click the "X" button to empty the Data files field.
Click the "..." button to open a file selection dialog.
- Define the key column containing the unique keys used for merging
identical feature rows from the multple data files.
The key column MUST be identical for all data files to merge.
- Number of feature (row) annotation columns.
These column will not be include into the merged table.
- Header row number: These rows will not be included into the merged table.
Apart from 1st row, which is used as column names in the merged matrix.
In a first step, SeedScan extracts all unique keys (ClonTracer barcode sequences) from all defined data files.
Next, matching data lines from all selected data matrices are placed into the merged matrix.
Missing records from a single data file are replaced with "0" cells.
.
The final result file contains the columns:
- Line counter
- ClonTracer Barcode sequence
- Row sum
- all count columns from the selected files
The final result file contains the rows:
- Names of the data columns found in the individual data files (1. row)
- Column sums
- All count columns from the selected files